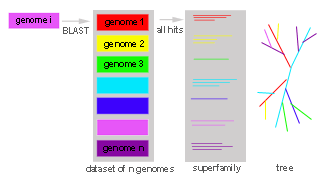
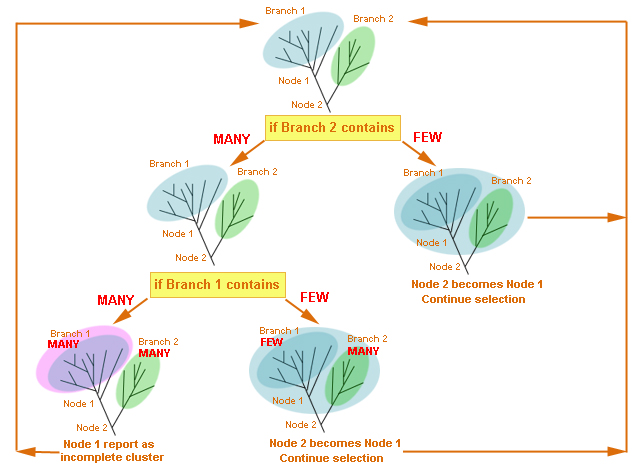
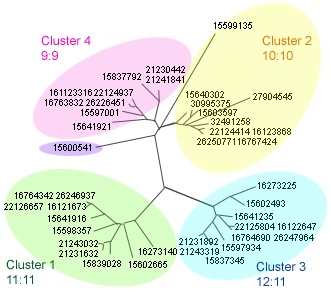
|
When a tree containing several clusters (i.e., families of orthologs) is
submitted to the BranchClust algorithm it is arbitrarily rooted: it can be
rooted inside any cluster or anywhere in between the clusters (see Figure
3). If the tree is rooted inside some cluster, the results of the clustering
will be affected by the root position; moreover, the undesirable split
inside a cluster containing the root could occur. For example, if the root
divides a cluster so that one of the resulting branches will contain more
than MANY species, this branch will be wrongly reported as a cluster.
However, if a tree is rooted somewhere in between the clusters, the results
will not be affected by the root position because a branch with a cluster
acts as a “stopper” in the algorithm, and no split inside a cluster will
occur.
The process of selection with tree re-rooting is depicted on Figure 3.
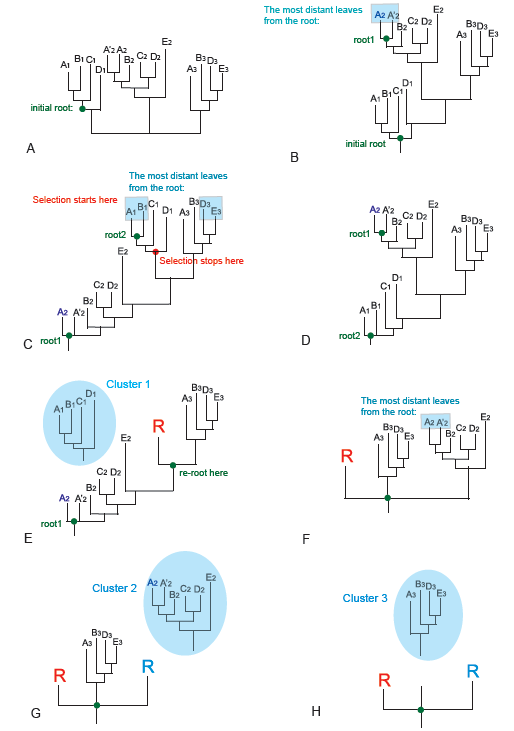
Figure 3.
BranchClust selection steps for superfamily of five taxa: A, B, C, D and E.
Figure 3A shows the original hypothetical unrooted tree for a set of 5
different taxa A, B, C, D and E. Let us set the parameter MANY to 4 (i.e.
the branch containing 4 different taxa will serve as a “stopper”) and let us
assume that the tree initially is arbitrarily rooted somewhere inside a
cluster – see green point in Figure 3A. Figure 3B shows the original tree
rooted at the green point inside a cluster.
We will run the algorithm twice with two different roots, which are chosen
as the two nodes most distant from each other in a tree. The process of root
selection for two independent runs is shown on Figures 3B-D. The first root,
root 1, is the ancestor of the most distant leaf in the initially rooted
tree (Figure 3B). The second root, root 2, is the ancestor of the most
distant leaf when the tree is rooted with the root 1 (Figure 3C). We run the
selection first for the tree rooted with root 1 (Figure 3C) and then with
root 2 (Figure 3D).
Figures 3C, E-H show how BranchClust works for the tree rooted with the
first root. According to the algorithm, we will start selection from the
most distant leaf which is either A1, or B1, or D3, or E3 (Figure 3C). In
case of more than two equally distant leaves we choose the first leaf. Then
we will descend node by node until we encounter a “stopper” - either a
branch containing MANY different species on condition that current branch
already contains MANY species, or a single leaf “R” signifying the removed
cluster. In our example selection stops at the red point because the next
node contains a branch with MANY (here 4) taxa (Figure 3C). The selected
cluster is removed from a tree (Figure 6E), the node is marked with leaf
“R”, the tree is re-rooted at the point of the cut, and then we repeat the
cycle for the re-rooted tree depicted on Figure 3F. Figure 3G shows how the
second cluster is selected and removed. There is no need to re-root the tree
if the node’s ancestor is already a root itself. Finally we have a tree
containing an only cluster (Figure 6H) which is selected at the third run of
the cycle.
We repeat selection for the tree rooted with the root 2 (Figure 6D) and
compare the results by calculating the number of paralogs resulting in two
different runs. The clustering that contains the least number of paralogs is
selected. Using two differently rooted trees helps to solve the problem that
arises in case of two incomplete clusters. This approach is illustrated by
the clustering of penicillin binding proteins’ superfamily for a set of 13
gamma proteobacteria (See Starting Point).
|