

- Open the Runtime menu at the top, and click on Restart Session: See A in the snapshot at right.
- Open the Edit menu at the top left, and click on Clear all outputs: See See B in the snapshot at right.
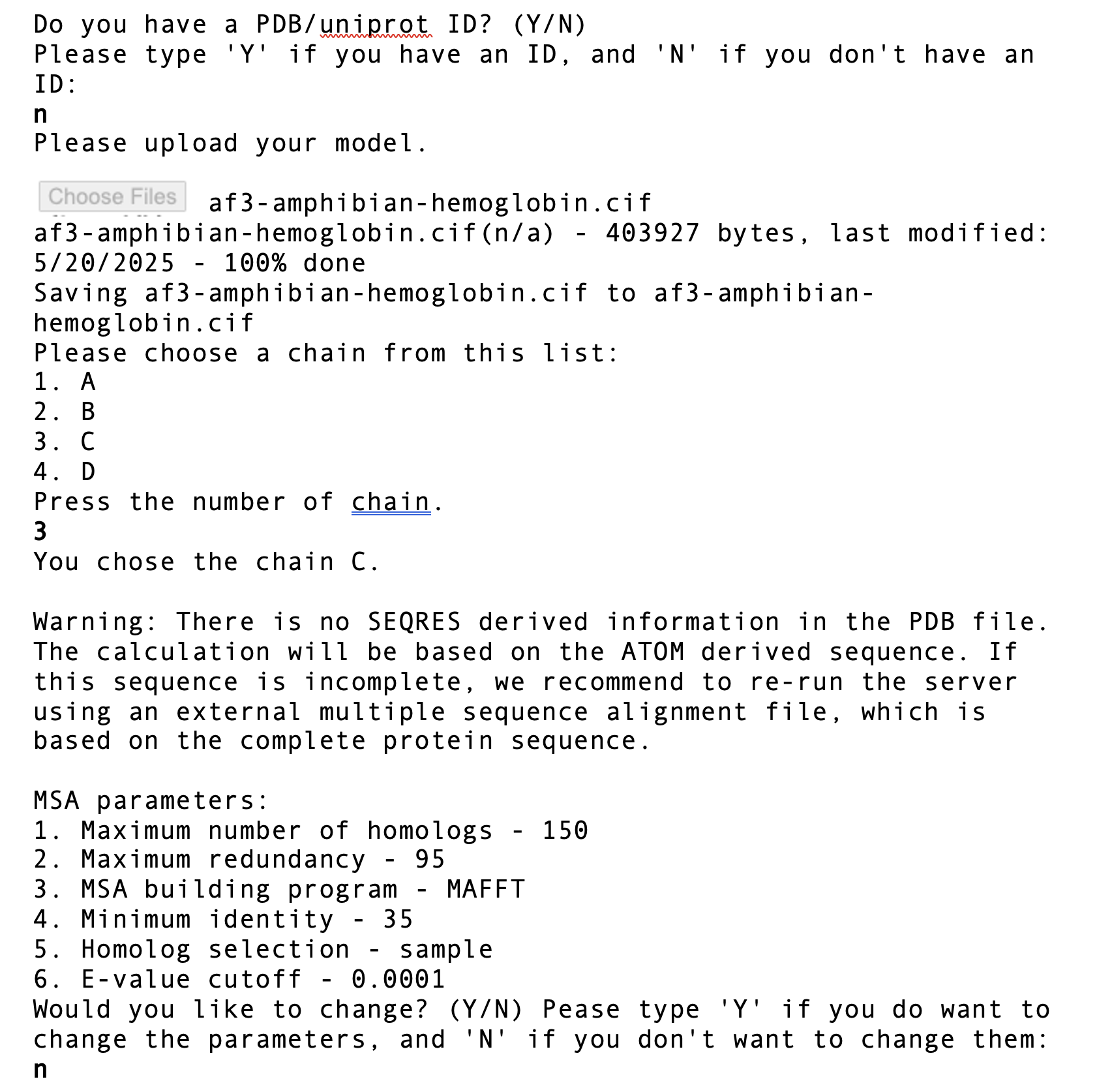
ConSurf is uniquely powerful in indicating when the multiple protein sequence alignment does not have sufficient information to calculate a meaningful conservation value for a given amino acid. When the confidence interval for the conservation score is excessive, the calculated conservation score for that residue is meaningless, and it is given conservation grade 10: insufficient data. This is obviously important to know. The only acceptable reason for hiding which amino acids have insufficient information would be if they are few, and not relevant to the point being made. "Hiding" means that the meaningless conservation grades for those amino acids will be shown. This can be accomplished by downloading a separate ConSurf result PDB or CIF file that omits insufficient data grades. When hidden, that fact must be disclosed.
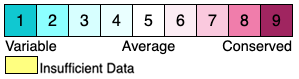
Conservation grades assigned by ConSurf are integers from 1 (highly variable) to 9 (highly conserved), with 10 (or 0) indicating insufficient data. The original ConSurf color scheme is below. Two alternative color schemes are also offered by FirstGlance when viewing ConSurf Colab results.
- Visualizing Conservation with FirstGlance.
- Interpreting ConSurf Results.
- Index to articles about ConSurf including Introduction to Evolutionary Conservation.
- Index to articles about FirstGlance.

The following links display ConSurf Colab-processed PDB files in FirstGlance:
- SV40 capsid:
1sva-1-consurf-colab.pdb.
To get the full capsid from the initial view, click on
I
Functional Assembly, then
II
360 Chains. Then depress the Slab button (Views tab), and check both checkboxes
in the Slab dialog to see the half-capsid. Which is more conserved, the inside or outside
surfaces of the capsid?
- Antibody (Immunoglobulin G):
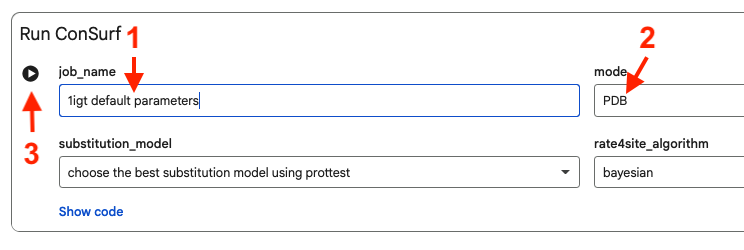
1igt-B-consurf-colab.pdb.
In the
Molecule Information Tab, under
Ligands+ & Non-Standard Residues, click on Find/List/See 2 covalent
glycoprotein bonds. How conserved are the glycosylated Asn residues?
- Bacteriophage T4 Cell Wall Puncturing Device:
Best first to look at the structure without ConSurf processing:
1k28. Try the Solid View.
Next: ConSurf-processed chain A: 1k28-A-consurf-colab.pdb. Again, follow steps I and II to get the functional assembly (biological unit 1).