|
At right is a fragment of a polypeptide chain. In the center is a single complete alanine residue. Check Alanine to identify its atoms1. The other atoms are fragments of adjacent amino acids2. Drag with your mouse to rotate the model. The Alanine is covalently bonded to other amino acids through peptide bonds. Check Peptide Bonds to locate them. The double bonds between main chain (backbone) C and O delocalize, making the peptide bonds also have partial double bonds (half-dotted bonds). This prevents the peptide bond from rotating. Each peptide bond holds six atoms in a plane. Check Planes to see them. The alpha carbon (Cα) in the center of each amino acid is held in the main chain by two rotatable bonds. The dihedral (torsion) angles of these bonds are called3 Phi and Psi (in Greek letters, φ and ψ). Use the radio buttons (top of right panel) to identify the rotatable main-chain bonds, and click the -20° and +20° buttons to see them rotate. Click the Reset button. The balls shown are much smaller than the atoms they represent. Check van der Waals to see the real sizes of the atoms4. In fact, most Phi and Psi angle combinations are impossible because two atoms cannot occupy the same space. Check Show Clashes to see where non-bonded atoms are overlapping, and thus in physically impossible positions. (This model simulation allows two atoms to overlap, unlike real atoms.) Check White to make clashes easier to see. Rotate Phi and Psi to find angle combinations where there are no clashes. In the early 1960’s, G. N. Ramachandran (University of Madras, India) and coworkers computationally determined the phi and psi angles that avoid steric collisions, initially treating the atoms simply as rigid spheres5, 6. They showed that the physically allowed angle combinations (that avoid clashes) correspond largely to the secondary structures observed in proteins: alpha helices, beta sheets, and turns. Ramachandran and team also showed that the major effect of sidechains on the allowed phi and psi angles is due to Cβ 2. Sidechains larger than that of alanine affect the allowed angles by only a few percent 7, 8. The Ramachandran Plot below shows the phi and psi angles actually observed in proteins. |
| |||||||||
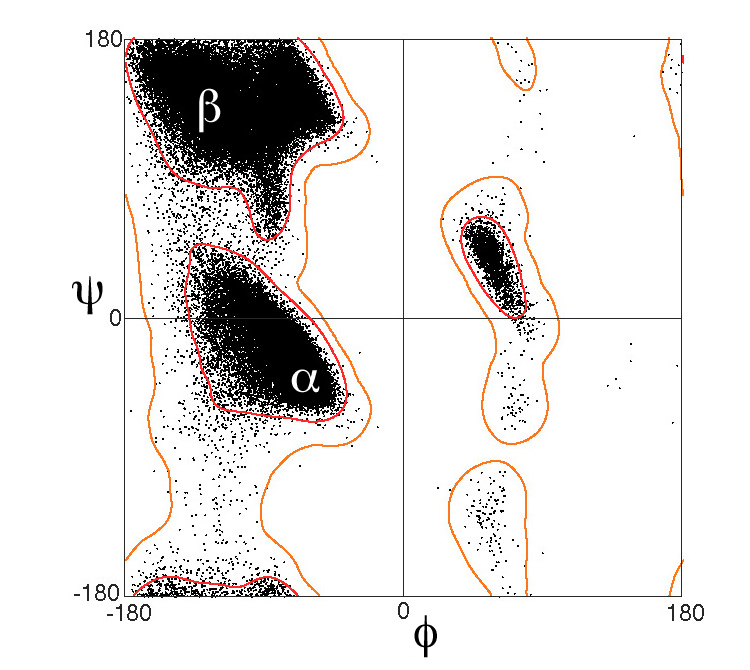
This plot excludes glycine (whose sidechain is a single hydrogen), proline (whose sidechain is covalently linked back to the main chain), and amino acids that precede proline. These special cases have different distributions on Ramachandran plots.
Challenge your understanding with the PRACTICE QUIZ.
- Dihedral (torsion) angles are explained with animated models rotating clockwise and counter-clockwise in the Slideshow and the YouTube Video.
- There is also a simple visualization of phi and psi angles at Dihedral angles in proteins by Angel Herráez.
- Tutorial: Ramachandran Plot Inspection: an interactive Ramachandran plot with many controls and details, by Angel Herráez. Also in Spanish.
- Ramachandran Plot: detailed explanation with example proteins and their plots displayed in JSmol. Here you can show the Ramachandran plot for any protein structure.
- A list of all related resources in English and Spanish: Dihedral/Index.
- Backbone representations explains the relations between backbone traces and main chain polypeptide bonds, as well as smoothed traces and ribbons.
|
This tutorial is available in two locations:
Proteopedia.Org
and
Bioinformatics.Org.
There is also a Slideshow, a YouTube Video, and a Practice Quiz. |
Notes & References
- The outlines of the black dots that identify the atoms in Alanine are smaller than the actual (van der Waals) sizes of those atoms. Check van der Waals to see the actual sizes.
- Each amino acid contributes 3 atoms directly to the main chain (backbone) of covalent bonds: -N-Cα-C The model here includes -C-C-N-C-C-N-C-. The central Cα has alanine's sidechain, -CH3. Alanine's sidechain carbon is termed Cβ.
- Edsall JT, Flory PJ, Kendrew JC, Liquori AM, Nemethy G, Ramachandran GN, Scheraga HA. A proposal of standard conventions and nomenclature for the description of polypeptide conformation. J Biol Chem. 1966 Feb 25;241(4):1004-8. PMID:5905118
- Actually, the van der Waals checkbox shows the atoms at 88% of their true van der Waals radii. In the above simulation, clashes are reported when 88% of the true radii overlap. This is in accord with the observations of Ramachandran and Sasisekharan6, who found that allowed interatomic distances for non-bonded atoms are ~0.4 Å less than their van der Waals radii10. The van der Waals radius of carbon is 1.7 Å. Thus, the van der Waals distance between the centers of two non-bonded carbon atoms is 3.4 Å. However the minimum allowed distance is about 0.4 Å less, which is 12% less. Thus 88% of the true van der Waals radii was used in the above simulation for detection of "clashes".
- Ramachandran, G. N., Ramakrishnan, C., Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J Mol Biol. 1963 Jul;7:95-9. PMID:13990617
- Ramachandran, G. N., Sasisekharan V. Conformation of polypeptides and proteins. Adv Protein Chem. 1968;23:283-438. PMID:4882249
- Ramakrishnan, C., Ramachandran, G. N. Stereochemical criteria for polypeptide and protein chain conformations. II. Allowed conformations for a pair of peptide units. Biophys J. 1965 Nov;5(6):909-33. PMID:5884016
- Chakrabarti P, Pal D. The interrelationships of side-chain and main-chain conformations in proteins. Prog Biophys Mol Biol. 2001;76(1-2):1-102. PMID:11389934
- The plot shown11 is available in the Wikimedia Commons courtesy of Jane and David Richardson.
- Ramachandran and Sasisekharan6 determined inter-atomic distances of closest approach of non-bonded atoms from crystal structures. For each pair of elements (their Table VI), they determined an allowed distance, and a partially allowed distance. Distances less than the partially allowed values are “very unlikely” to occur due to steric repulsion. The allowed distances are 0.3 to 0.5 Å less than the van der Waals radii. (their page 327). The partially allowed distances are usually 0.1 Å, sometimes 0.2 Å, less than the allowed distances.
- Lovell SC, Davis IW, Arendall WB 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by C-alpha geometry: phi, psi and C-beta deviation. Proteins. 2003 Feb 15;50(3):437-50. PMID:12557186
This page is by Eric Martz.
License: Attribution-NonCommercial-ShareAlike 4.0 International.
Released May 27, 2018. Enhanced June 24 and July 16, 2018.
Many thanks to Bob Hanson and the JSmol Team, and to Jaime Prilusky for adaptation to Proteopedia.Org.