![]()
A step-by-step guide on how to download complete genomes, assemble superfamilies, reconstruct trees, select orthologous families with BranchClust, and analyze and depict results with TreeDyn.
|
Perl Script Home |
| Perl-script only: |
|
the latest version: version 1.01 November, 2006: BranchClust_1-01.pl previous versions: version 1.00 June, 2006: BranchClust_1-00.pl |
| Perl-script and examples: |
contains the perl-script together with the examples of trees and various taxa recognition files.
| BranchClust Tutorial: |
|
|
A step-by-step guide on how to download complete genomes, assemble superfamilies, reconstruct trees, select orthologous families with BranchClust, and analyze and depict results with TreeDyn. |
contains all the perl scripts described in the Tutorial (updated January 19, 2008)
| Program Usage |
Required:
| 1. |
Bioperl module for parsing trees Bio::TreeIO. For instructions how to install bioperl go here. |
|
2.
|
Taxa recognition file gi_numbers.out must be present in the current directory. How to create this file, read the Taxa recognition file section. |
A
t the command line type:|
# perl BranchClust.pl <tree-file> <MANY> |
|
where <tree-file> is a superfamiliy tree in PHYLIP format, and <MANY> is a parameter designating the minimum number of different taxa on the branch sufficient for the branch to be a separate cluster (see Algorithm). It can be any number less or equal to the number of different taxa considered. The BranchClust algorithm usually works well with the value of 80% of total number of different taxa. See Examples below. |
| Output |
|
BranchClust
creates three output files:
clusters.out,
families.list
and
cluster.log.
File
clusters.out
reports clusters with selected families and IN- and OUT-OF-CLUSTER paralogs,
if any. File
families.list
contains only the list of selected families. Log-file
cluster.log
tracks selection algorithm, as it parses trees, node by node, and can be useful
to analyze
cases of wrong clustering. |
| Taxa recognition file |
|
The structure of taxa recognition file gi_numbers.out is defined as follows:
species1 | match_pattern1.1 match_pattern1.2 .. match_pattern1.m species2 | match_pattern2.1 match_pattern2.2 .. match_pattern2.m species3 | match_pattern3.1 match_pattern3.2 .. match_pattern3.m ... speciesN | match_patternN.1 match_patternN.2 ... match_patternN.m
where species1, species2, species 3 are different taxa and match_patterns are the ordinary perl pattern-matching expressions that can be used as an unique identifier of taxa. Species names are for user convinience only, and everything preceding sign | is ignored. Match patterns can be either names or gi_numbers. |
|
1. Example of how to use names as taxa identifiers: |
|
Suppose we have 4 different taxa, and we need to analyze a tree reconstructed from a mixture of paralogs and orthologs:
The taxa recognition file could be as follows:
species1| bacteria1.* species2| bacteria2.* species3| bacteria3.* species4| bacteria4.*
The paralogs of the same taxa can be designated as bacteria1.1, bacteria1.2, bacteria2.1, bacteria2.2, etc.
Here is example of tree containing paralogs and orthologs of 4 different taxa:
(((bacteria1_01,bacteria2_01),(bacteria3_01,bacteria4_01)),(bacteria3_03, (bacteria3_02,(bacteria2_02,(bacteria1_02,bacteria1_03)))));
How does BranchClust parse this tree, see Examples below. |
|
2. Example of how to use gi-numbers as taxa identifiers: |
|
Suppose we have 4 species of bacteria and archaea. The species could be distinguished by their gi-numbers:
Bacillus subtilis subsp. subtilis str. 168 | 1607.... 5081.... 1608.... 1867.... Escherichia
coli K12 | 1612.... 4917.... 1613.... 3334....
Methanosarcina mazei Go1 | 2122.... Sulfolobus
solfataricus P2 | 1589.... Here is the tree for the superfamily of ATP-ases (see Algorithm and Tutorial to learn how to assemble superfamilies), with gi_numbers to identify genes.
((((16078687:0.38341,16129888:0.51097):0.17454,(16080761:0.29985, 16131639:0.32318):1.63425):0.29299,((16080736:0.26753,16131602:0.39301):0.70160, (15897485:0.22830,21226881:0.36977):0.81741):0.26223):0.20986,(16131600:0.17847, 16080734:0.27503):0.66717,21226882:1.19107);
How does BranchClust parse this tree, see Examples below. |
|
3. File with gi numbers identifiers for 319 bacterial and archaeal species: |
|
gi_numbers.out (created from genomes downloaded from ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria on April 6, 2006) |
|
|
Sample superfamily for 4 bacteria
# perl branch_clust.pl tree.tre 3 results: clusters.out, families.list
|
|
|
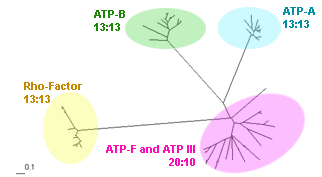
Superfamily of ATP synthases for 13 gamma proteobacteria
# perl branch_clust.pl tree.tre 8 results: clusters.out, families.list |
|
|
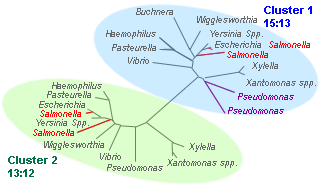
Superfamily of ATP synthases for 30 taxa (16 bacteria and 14 archaea)
# perl branch_clust.pl tree.tre 7 results: clusters.out, families.list |
|
|
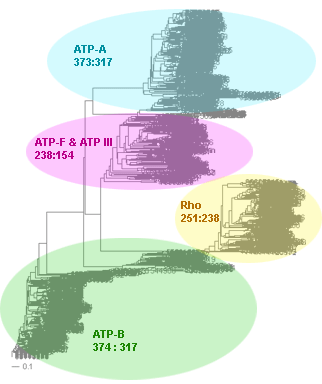
Superfamily
of ATP synthases for 317 taxa (bacteria and archaea)
# perl branch_clust.pl tree.tre 150 results: clusters.out, families.list
|
|
|
Superfamily of penicillin-binding proteins for 13 gamma proteobacteria
# perl branch_clust.pl tree.tre 5 results: clusters.out, families.list |
| How to do batch processing |
| Example of a wrapper you can find here: branch_clust_wrapper.pl |
| Coloring with TreeDyn |
Go to TreeDyn web-site to learn how to create annotation and script files.
Example for Superfamily of ATP synthases for 30 taxa (16 bacteria and 14 archaea)
|
|
Annotation file: labelfile.tlf Script file: script.tds Resulting tree: tree.png
|
|
Links |
Gogarten Lab Home Page: http://gogarten.uconn.edu/
Email to: Maria.Poptsova@uconn.edu
Page last updated: January 19, 2008